L'HEMOSTASE
Source : Association pour le Développement de l’Hématologie et de
la Transfusion
(Plus d’infos encore sur le site http://www.adhet.org
)
INTRODUCTION
L'hémostase est l'ensemble des mécanismes qui concourent à maintenir le
sang à l'état fluide à l'intérieur des vaisseaux.
Le processus d'hémostase, qui vise donc à arrêter les hémorragies et empêcher les thromboses se déroule classiquement en trois temps :
- l'hémostase primaire qui ferme la brèche vasculaire par un "thrombus blanc" (clou plaquet-taire),
- la coagulation qui consolide ce premier thrombus en formant un réseau de fibrine emprisonnant des globules rouges (thrombus rouge),
- la fibrinolyse, processus limitant, permettant la destruction des caillots, ou la limitation de leur extension.
Ces trois temps sont initiés simultanément dès qu'est enclenché le processus d'hémostase.
I - HEMOSTASE PRIMAIRE
Immédiatement déclenchée dès qu'il y a une brèche vasculaire, elle aboutit à l'arrêt du saignement essentiellement pour les petits vaisseaux.
A - LES ACTEURS EN PRESENCE
Quatre éléments principaux sont impliqués dans l'hémostase primaire :
- deux éléments cellulaires : cellules endothéliales et plaquettes
- deux éléments plasmatiques : facteur von Willebrand et fibrinogène
1 - Endothélium et paroi vasculaire
Toutes les parois vasculaires de l'organisme sont construites sur un schéma identique comportant trois couches (cf. schéma) :
- l'intima qui est faite d'une couche continue monocellulaire de cellules endothéliales séparée de la substance sous endothéliale par la membrane basale.
Les cellules endothéliales
tapissent l'ensemble des vaisseaux. Elles ont des fonctions multiples. En
s'interposant de façon ininterrompue entre le sang et les substances
sous-endothéliales procoagulantes, les cellules endothéliales préviennent
l'activation de la coagulation et des plaquettes. Elles constituent en outre un
filtre sélectif puisque des cellules et des substances diverses peuvent sortir
de l'endothélium en passant entre les cellules endothéliales, d'autres
substances peuvent transiter à travers la cellule endothéliale par des réseaux
de canaux par un mécanisme de pinocytose.

Fig
1: Structure de la paroi vasculaire
En outre, à l'état de repos, les cellules endothéliales sont antithrombotiques. Par contre lorsqu'elles sont activées, elles deviennent prothrombotiques et seront le support des réactions aboutissant à la coagulation du sang. Enfin ces cellules sont douées de propriétés de synthèse extrêmement importantes : elles synthétisent des facteurs impliqués dans l'hémostase : facteur Willebrand, prostacycline (PGI2), facteur tissulaire, thrombomoduline, activateur du plasminogène (tPA) et son inhibiteur (PAI).
Les cellules endothéliales sont impliquées dans des phénomènes autres que l'hémostase: phénomènes immunitaires (cellules présentatrices de l'antigène, synthèse de facteur de croissance hématopoïétique...).
- la membrane basale : elle est faite de collagène, les cellules endothéliales reposent dessus. La membrane basale joue le rôle de "tamis" moléculaire.
- le sous-endothélium comporte des microfibrilles constituées d'un autre type de collagène. Il est très thrombogène.
L'intima est séparée de la média par la limitante élastique interne.
- Média : elle est plus ou moins développée suivant les vaisseaux : l'artère comporte une média importante. La média est riche en fibres musculaires qui permettent la vasoconstriction et en fibroblastes.
La média est séparée de l'adventice par la limitante élastique externe.
- L'adventice fera le lien avec les autres structures tissulaires péri vasculaires. Dans l'adventice circulent les vasa vasorum et se terminent les ramifications nerveuses.
2 - Plaquettes
Les plaquettes sont les plus petits éléments figurés du sang. De l'extérieur vers l'intérieur elles comportent (Fig.2A) :
- une membrane composée d'une double couche de phospholipides (PL) s'opposant par leur pôle hydrophobe (Fig.2B). Une propriété importante de la membrane est la répartition asymétrique des PL notamment de la phosphatidylsérine (PS). On se rappellera aussi de la richesse en acide arachidonique de la membrane. Dans cette membrane sont implantées des glycoprotéines dont les principales sont la glycoprotéine IIb IIIa et la glycoprotéine Ib. Cette membrane est riche en récepteurs divers, le plus important dans la physiologie plaquettaire étant le récepteur à la thrombine récemment individualisé.

Fig 2B: La membrane plaquettaire
Double couche de phospholipides et glycoprotéines
Sous la membrane plaquettaire on trouve un réseau musculo-squelettique fait de micro fibrilles composées d'actine et de myosine qui constituent une véritable musculature pour la plaquette douée de mouvements propres et un squelette fait de micro tubules qui contribuent à maintenir la forme discoïde de la plaquette.
A l'intérieur des plaquettes on trouve, dans le cytoplasme, deux réseaux de canaux :
. le système canaliculaire ouvert, fait de profondes invaginations dans la membrane plaquettaire, permettant une communication rapide entre des éléments extra cellulaires et l'intérieur des cellules
. le système tubulaire dense, lieu de stockage du calcium
Dans le cytoplasme on reconnaît des granulations de trois types :
. granules denses comportant de l'ATP, de l'ADP, de la sérotonine et du calcium,
. granules alpha comportant du facteur 4 plaquettaire, de la beta thromboglobuline, du facteur Willebrand et de très nombreuses autres substances,
. grains lysosomiaux faits d'enzymes très divers (hydrolases, phosphatases).
Enfin on trouve dans la plaquette des grains de glycogène et des mitochondries.
3 - facteur von Willebrand (vWF)
Il s'agit d'un polymère hétérogène composé de multimères de poids variable (0,5 à 15 x 10 6 Daltons). Le facteur Willebrand est synthétisé par les cellules endothéliales et les mégacaryocytes. Il est présent dans le plasma et les plaquettes. Dans le plasma, il circule lié au facteur antihémophilique A (facteur VIII). Le facteur Willebrand protège le facteur VIII, qui est un facteur labile, contre la protéolyse. Une diminution importante du facteur Willebrand entraînera donc une diminution du facteur VIII.
4 - fibrinogène
Cette molécule est un dimère. Chaque monomère est composé de trois chaînes (alpha, bêta, gamma). La molécule du fibrinogène comporte un domaine central E et deux domaines latéraux D (voir p. 86). Le fibrinogène interviendra dans l'hémostase primaire mais aussi dans la coagulation.
B - LE DEROULEMENT DE L'HEMOSTASE PRIMAIRE
Dès qu'une brèche vasculaire se constitue, le processus d'hémostase primaire se met en jeu. Il comprend :
1 - Le temps vasculaire
La première réaction de l'organisme est une vasoconstriction localisée qui peut soit arrêter les hémorragies, soit au moins réduire le flux sanguin et modifier les conditions hémodynamiques, favorisant le processus d'hémostase.
2 - L'adhésion plaquettaire
Les plaquettes dès leur sortie du vaisseau adhèrent à la structure sous endothéliale mise à nu par la brèche vasculaire. L'adhésion se produit en grande partie par la glycoprotéine Ib qui se colle au sous endothélium grâce au facteur Willebrand qui sert de ciment. Une première couche monocellulaire de plaquettes se constitue ainsi. Les plaquettes adhérentes s'activent et recrutent d'autres plaquettes circulantes.
3 - L'agrégation plaquettaire
Sur la première couche de plaquettes se fixent d'autres plaquettes, par des phénomènes de membrane : l'agrégation plaquettaire se fait grâce au fibrinogène qui établit un pont entre les plaquettes par l'intermédiaire des glycoprotéines IIbIIIa présentes à la surface des plaquettes activées. Ce phénomène d'agrégation, extensif crée un premier thrombus fragile. On dit que l'agrégation est réversible. Grâce à la libération des enzymes et du contenu granulaire des plaquettes, le caillot se solidifie : on parle d'agrégation irréversible, ce qui va constituer le thrombus blanc ou clou plaquettaire (Fig. 3A et 3B).
4 - Les fonctions procoagulantes plaquettaire
La phosphatidylsérine (PS) est un phospholipide localisé à l'intérieur de la membrane plaquettaire au repos. Après activation, la PS est externalisée. Elle va servir de support et de surface de catalyse aux réactions de coagulation.
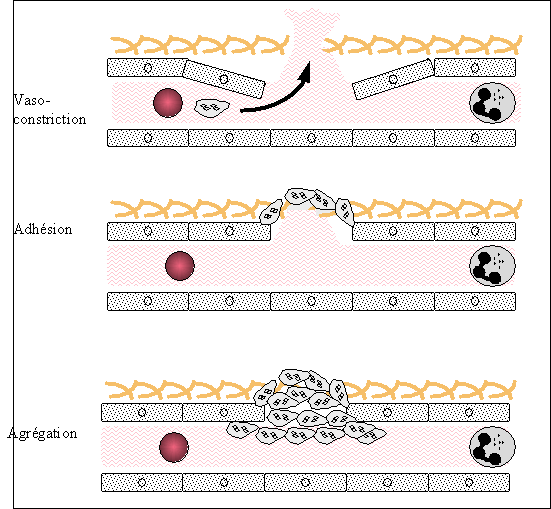
Fig 3A: Déroulement de l’hémostase primaire
II - COAGULATION
Le thrombus plaquettaire est fragile. Il sera renforcé par l'apparition concomitante d'un autre thrombus, le thrombus rouge résultat de la coagulation.
1 - Schéma global de la
coagulation
La coagulation est la gélification du plasma. Dans le plasma circule une substance soluble, le fibrinogène, déjà évoqué. Le processus de coagulation permet la transformation du fibrinogène soluble en un gel de fibrine insoluble qui obture la brèche vasculaire et consolide le caillot.
La coagulation est une cascade de réactions enzymatiques. L'enzyme qui permettra de transformer le fibrinogène en fibrine est la thrombine. Son processus de formation est complexe. Il comprend une série ordonnée d'activations enzymatiques qui surviennent à la surface des PL membranaires de certaines cellules (plaquettes, cellules endothéliales, monocytes). Dans le plasma circulent des proenzymes, inactifs, appelés facteurs de coagulation (exemple : facteur X, facteur VII). Lorsque ces pro enzymes sont activés on parle de facteurs de coagulation activés (exemple : facteur Xa, VIIa). Deux facteurs de la coagulation sont dépourvus d'activité enzymatique, le facteur VIII et facteur V. Ce sont des cofacteurs qui augmentent la vitesse d'activation des autres enzymes. La liste des facteurs de coagulation figure dans le tableau I.
2 - Voie extrinsèque- voie intrinsèque
On décrit classiquement deux voies d'activation de la coagulation, la voie extrinsèque et la voie intrinsèque qui se rejoignent au niveau de l'activation du facteur X.
.
voie intrinsèque
Intervention du système contact : ce système comprend quatre facteurs qui sont le facteur XII, le facteur XI, la prékallikréine et le kininogène de haut poids moléculaire. L'activation du système contact peut être déclenchée par le contact du facteur XII avec une surface chargée négativement mouillable ou certains composés biochimiques (complexes immuns par exemple).
Le complexe antihémophilique comprend deux facteurs extrêmement importants en pathologie, le facteur IX ou facteur antihémophilique B et le facteur VIII ou facteur antihémophilique A.
Le facteur IX activé en présence de facteur VIII activé qui est son cofacteur permet l'activation du facteur X en facteur X activé (ce que nous reverrons plus loin).
.
voie extrinsèque (Fig.4)
Cette voie nécessite la présence d'une protéine non enzymatique, appelée facteur tissulaire (FT). Ce composé protéique est présent dans beaucoup de tissus. Certains tissus sont très riches en facteur tissulaire : tissu cérébral par exemple. In vivo, le facteur tissulaire est exprimé à la surface de certaines cellules ; le FT ne circule pas dans le plasma. Cellules endothéliales, monocytes, quand ces cellules sont activées, notamment par des toxines bactériennes ou des cytokines (IL1, TNF), le FT est exprimé au niveau des cellules musculaires lisses de la paroi vasculaire et des fibroblastes de façon constitutive.
En présence de facteur tissulaire lié aux phospholipides des membranes cellulaires, le facteur VII ou proconvertine s'active et devient la convertine, ou facteur VIIa. Le facteur VIIa lié au facteur tissulaire permet lui aussi d'activer le facteur X ou facteur Stuart en facteur X activé.
Dans les deux cas, la voie finale commune comprend l'activation du
facteur X en facteur Xa.
3 - la thrombinoformation
Que la coagulation ait été activée par voie extrinsèque ou intrinsèque, un facteur important est généré: le facteur X.
Le facteur X activé s'intégrera dans un complexe appelé prothrombinase qui comprend outre le facteur X activé, le facteur V activé, les phospholipides de la surface cellulaire et du calcium.
Le complexe prothrombinase protéolyse le facteur II ou prothrombine et forme la thrombine.
La thrombine (facteur IIa) est une enzyme extrêmement puissante, c'est elle qui va coaguler le fibrinogène. Une molécule de thrombine peut coaguler 1000 fois son poids de fibrinogène. En outre, la thrombine catalyse sa propre formation puisque c'est la thrombine qui active le facteur VIII en facteur VIII activé et le facteur V en facteur V activé.

Fig 4 : Le rôle du facteur tissulaire
La fixation du facteur VII au facteur tissulaire permet l’activation du facteur VII. Le facteur VII activé peut soit activer directement le facteur X (si le facteur tissulaire est en excès), soit activer le facteur IX qui en présence de facteur VIII activera le facteur X. (fig. 4).
4 - la fibrinoformation
L'action de la thrombine sur le fibrinogène libère deux petits peptides, appelés fibrinopeptides A et B. Il ne reste alors que des monomères de fibrine qui spontanément se polymérisent. Ce premier polymère de fibrine est instable, soluble dans l'urée: on parle de polymères solubles. Il va être stabilisé par un dernier facteur, le facteur XIII (facteur stabilisant la fibrine: FSF). Le facteur XIII va créer des liaisons covalentes solides entre ces monomères de fibrine. On a alors formation d'un réseau de fibrine qui emprisonne les globules rouges : le thrombus rouge définitif est ainsi formé.

Fig 5: La coagulation in vivo
Quelle que soit la voie d’activation, le facteur X en présence de facteur Va active la prothrombine (facteur II) en thrombine qui permettra la transformation du fibrinogène en fibrine, structure de base du caillot
5 - La coagulation in
vivo/in vitro (Fig.5)
Quelques éléments sont à souligner pour compléter ce schéma de la coagulation :
a) Voie extrinsèque, voie intrinsèque: quelle
est la voie prépondérante ?
Des travaux récemment publiés suggèrent que l'activation de la coagulation par les voies extrinsèque et intrinsèque ne correspond pas aux mécanismes in vivo. La voie du FT est prépondérante in vivo. La conception des 2 voies reste utile pour l'exploration car la voie endogène et la voie exogène sont respectivement explorées par le temps de céphaline activée et le temps de Quick.
b) rôle du calcium
Cet électrolyte est indispensable à la coagulation. Seule l'activation du système contact peut se faire en son absence.
Cette propriété est utilisée lors des prélèvements sanguins : si l'on veut éviter la coagulation du sang, il suffit de prélever sur un chélateur du calcium (EDTA, citrate). On peut ensuite soit laisser sédimenter les globules rouges ou ce qui est plus rapide centrifuger le prélèvement ; on sépare alors les cellules du plasma, c'est cette méthode qui est utilisée en pratique quotidienne. Le plasma par définition comprend tous les facteurs de coagulation. Les tests de coagulation sont réalisés sur le plasma.
Par contre en présence de calcium et de surface mouillable, le sang coagule. Si l’on élimine le caillot, il reste non plus du plasma mais du sérum : le sérum diffère du plasma par l'absence de certains facteurs de coagulation qui sont complètement consommés lors de la coagulation. Les facteurs consommés sont le fibrinogène (facteur I), le facteur V et le facteur VIII. Le sérum est incoagulable, il ne peut donc pas être utilisé pour faire des examens de coagulation (sauf le test de consommation de la prothrombine p. 88).
c) lieu de synthèse et métabolisme (Tableau I)
Tous les facteurs de coagulation sont synthétisés par l'hépatocyte, ceci explique les désordres hémorragiques chez les cirrhotiques ou les personnes atteintes d'une insuffisance hépato-cellulaire. Le facteur VIII fait exception à cette règle : son taux reste normal ou augmenté.
En outre, certains facteurs nécessitent pour être synthétisés la présence d'une vitamine : la vitamine K. Ces facteurs dits "vitamine K dépendant" sont les facteurs II, VII, X et IX (prothrombine, proconvertine, facteur Stuart, facteur antihémophilique B). On les désigne habituellement par PPSB du nom de leurs initiales. La vitamine K permet la carboxylation des facteurs PPSB, processus nécessaire à la fixation du Ca2+. Un patient porteur d'une avitaminose K ou recevant un traitement appelé antivitamine K aura une diminution de synthèse de ces quatre facteurs. A la place circulent des substances appelées PIVKA (Protein Induced by Vitamine K Absence ou Antagoniste) : PIVKA 7, PIVKA 2, PIVKA 10, PIVKA 9 : ce sont des précurseurs non carboxylés des facteurs PPSB. Elles sont inactives dans la coagulation. Leur liaison aux phospholipides en présence de calcium est impossible.
N° Facteur Nom Particularité Demi-vie Taux mini
nécessaire
Facteur I Fibrinogène Absent du sérum 4-6 jours 0,5 à 1 g/l
Facteur II Prothrombine Vit K dépendant 3-4 jours 40 %
< 5 % dans sérum
Facteur V Proaccélérine Absent du sérum 12-36 h 10-15 %
Facteur VII Proconvertine Vit K dépendant 4-6 h 5-10 %
Facteur VIII Anti-hémoph A Absent du sérum 10-16 h 30%
Facteur IX Anti-hémoph B Vit K dépendant 24 h 30%
Facteur X Stuart Vit K dépendant 1-2 jours 10-20%
Facteur XI Rosenthal 1-2 jours 30%
Facteur XII Hageman 2-3 jours 0% ?
Facteur XIII Stabilisant fibrine 3-7 jours 2%
Tableau I: Les facteurs de coagulation
6 - Régulation de la coagulation : le rôle des inhibiteurs (Fig. 6)
Le système de la coagulation plasmatique a tendance à s'activer spontanément. Il est très important pour l'organisme que les enzymes formés lors de l'activation de la coagulation (thrombine, facteur Xa) ne circulent pas dans le plasma car ils risqueraient d'entraîner une activation diffuse de la coagulation et un processus pathologique grave. Pour éviter ceci et maintenir leur équilibre, chaque facteur activé a son inhibiteur. On connaît trois systèmes inhibiteurs : le système de l'antithrombine, le système Protéine C-Protéine S, et le TFPI.
- l'antithrombine (anciennement appelée antithrombine III: ATIII) inhibe principalement le facteur II activé ou thrombine mais aussi le facteur X activé, le facteur IX activé et partiellement le facteur XI activé. Son activité anticoagulante est augmentée de façon très importante par un produit utilisé en thérapeutique, l'héparine.
Les déficits en antithrombine sont des maladies sévères responsables de thromboses à répétition (thrombose veineuse, embolie pulmonaire).
Il existe un autre inhibiteur de la thrombine mais qui a peu d'importance physiologique, semble-t’il : le second cofacteur de l'héparine.
-
le système Protéine C-Protéine S
La protéine C circule sous forme inactive. Elle peut être activée par la thrombine en Protéine C activée à condition que la thrombine soit fixée sur un récepteur appelé la thrombomoduline. La Protéine C activée est un inhibiteur très puissant des facteurs Va et VIIIa. Son action est augmentée par une autre substance circulant dans le sang, la Protéine S. Il est intéressant de noter que la Protéine C et la Protéine S sont des facteurs vitamine K dépendants.
Il existe des déficits en protéine C et S exposant les sujets atteints à un risque de thrombose.
Dans les substrats de la Protéine C, le plus important paraît être le facteur Va. Certains individus présentent une anomalie du facteur V qui rend le Facteur Va insensible à l'action neutralisante de la PCa : on parle de résistance à la Protéine C activée (RPCA). Cette anomalie est très fréquente (~ 3 % dans le Sud de la France). Elle est associée à une anomalie moléculaire sur le gène du facteur V appelé facteur V Leiden (ou mutation R506 Q). Les sujets ont un risque modérément augmenté de thromboses veineuses.
- le TFPI (tissue factor pathway inhibitor)
On a longtemps cherché quel pouvait être l'inhibiteur du facteur VII activé. Il n'y a pas d'inhibiteur du facteur VII activé mais un inhibiteur appelé TFPI qui inhibe l'activation du facteur X par le complexe [facteur VIIa-FT]. Ceci explique que, dans le plasma, circule un peu de facteur VII activé.

Fig 6: Les inhibiteurs de la coagulation
Il y a 3 systèmes inhibiteurs principaux:
l’antithrombine (ATIII), le système protéine S - protéine C
et l’inhibiteur de la voie du facteur tissulaire (TFPI)
III - LA FIBRINOLYSE
La fibrinolyse est le troisième temps de l'hémostase. Sa finalité est l'inverse de celles de l'hémostase primaire et de la coagulation qui visaient à former des caillots. La fibrinolyse tend à le détruire ou à l'empêcher de se former.
La fibrinolyse fait intervenir une substance circulant sous forme inactive dans le plasma: le plasminogène, synthétisé par le foie. Sous l'influence d'activateurs, le plasminogène se transforme en plasmine qui est une enzyme protéolytique très puissante, capable de dégrader le caillot de fibrine mais aussi de détruire le fibrinogène, voire d'autres facteurs de coagulation. La fibrinolyse est normalement un processus localisé au niveau du caillot. La génération voire la circulation de la plasmine doivent être régulées pour maintenir un équilibre physiologique.
Les voies d'activation du plasminogène sont au nombre de deux :
- la voie du t-PA (activateur tissulaire du plasminogène). Cette substance est synthétisée par la cellule endothéliale qui la libère lorsqu'elle est en état d'hypoxie, de stress ou lors de tout phénomène d'agression.
- la voie de la prourokinase-urokinase. La forme circulante est la pro-urokinase synthétisée par les cellules rénales et d'autres cellules parenchymateuses. La pro-urokinase s'active en urokinase au contact du caillot de fibrine. Il est intéressant de noter que le système contact (facteur XII et kallicréine) peuvent activer la pro-urokinase.
Régulation :
Le système fibrinolytique est régulé par des inhibiteurs. On distingue deux types d'inhibiteurs :
- inhibiteurs de la plasmine : alpha 2 antiplasmine, alpha 2 macroglobuline
- inhibiteurs des activateurs du plasminogène : le PAI-1 est l'inhibiteur surtout du t-PA et le PAI-2, présent essentiellement chez la femme enceinte, est inhibiteur de l'urokinase. Il a été décrit aussi un PAI-3 et d'autres inhibiteurs de surface cellulaire.

Fig 8 : Coagulation et fibrinolyse
Les produits de dégradation de la fibrine et du fibrinogène.
La plasmine génère des fragments très hétérogènes à partir de la fibrine. Ces fragments portent tous la structure D-D d'où le nom de D-Dimères (Fig.7)
Synthèse
Le processus d'hémostase primaire et de coagulation aboutit à la formation d'un caillot alors que la fibrinolyse tend à le détruire. Il y a donc un équilibre permanent entre d'un côté l'hémostase primaire et la coagulation et d'un autre côté la fibrinolyse. Cet équilibre s'appelle balance coagulolytique (Fig. 8).
- Une hémorragie peut être due soit à un défaut de l'hémostase primaire (thrombopénie, thrombopathie), soit à une coagulopathie (absence d'un ou plusieurs facteurs de coagulation), soit à un excès de fibrinolyse (excès d'activation ou défaut d'inhibiteurs).
- Une thrombose peut être due à une activation excessive de la coagulation favorisée par un déficit des inhibiteurs de la coagulation. Théoriquement, les hypofibrinolyses peuvent être aussi responsables de thrombose. Récemment les excès de facteurs de la coagulation notamment de FVIII ont été décrits comme favorisant le risque de thrombose veineuse.
IV - EXPLORATION DE L'HEMOSTASE
L'étude de l'hémostase est extrêmement importante en clinique. Les tests d'hémostase sont utilisés : (1) pour le diagnostic d'un syndrome hémorragique, ou pour essayer de prévoir un risque hémorragique avant une intervention chirurgicale ; ou bien, (2) dans le cadre de thromboses à répétition, pour déterminer la cause de ces maladies invalidantes et graves puisque certaines peuvent entraîner la mort par embolie pulmonaire.
On ne dispose d'aucun test global d'étude de l'hémostase: on aura donc recours à des tests qui exploreront soit l'hémostase primaire, soit la coagulation, soit la fibrinolyse.
1 - Tests explorant l'hémostase primaire
a) Le temps de saignement
Le temps de saignement est le temps d'arrêt de l'hémorragie d'une plaie cutanée superficielle. Deux méthodes sont possibles :
- l'une consiste à faire une incision à l'oreille avec un vaccinostyle et à mesurer le temps d'arrêt du saignement. En moyenne il est de 2 à 4 min (test de Duke). Ce test ne doit plus être pratiqué.
- la seconde méthode consiste à mettre un garrot au bras gonflé à une pression de 4 cm de mercure et à faire une incision horizontale à l'avant-bras avec un appareil spécial jetable. Le saignement s'arrête en 4 à 8 min. Cette méthode s'appelle Ivy. C'est la plus sensible et la seule qui doit être utilisée en pratique.
Le temps de saignement apporte un certain nombre de renseignements mais il n'est pas infaillible. Il peut être perturbé par des erreurs techniques :
- incision trop profonde qui donne un faux allongement du temps de saignement,
- incision trop superficielle qui au contraire donne un temps trop court.
En outre le temps de saignement est allongé par la prise récente de certains médicaments en particulier l'aspirine. Il est donc indispensable de bien interroger les patients avant de faire un temps de saignement. Il peut être allongé dans les maladies de l'hémostase primaire (thrombopathie, thrombopénie, maladie de Willebrand).
En pratique cet examen est de moins en moins pratiqué. Il ne peut en aucun cas être considéré comme un examen de dépistage du risque hémorragique mais peut s'inscrire dans une démarche diagnostique à condition de bien en poser les indications.
b) La numération plaquettaire
Cet examen est capital. Il fait partie de tout bilan sanguin. Les appareils automatiques sont actuellement d'une grande reproductibilité. Les taux normaux de plaquettes sont de 150 à 400 x 109/l (150 à 400 000/ mm3 ou 150 à 400 – Giga/l)
Il faut savoir néanmoins qu'il existe de fausses thrombopénies lorsqu'il y a agrégation des plaquettes dans le tube, ce qui se produit lorsque le prélèvement a été mal fait (présence de micro caillots qui consomment des plaquettes). En outre, une circonstance à connaître est le fait que certaines personnes ont une agrégation des plaquettes en présence d'EDTA, anticoagulant utilisé sur les tubes à hémogramme.
Toute thrombopénie doit donc être vérifié sur un prélèvement effectué
sur tube citraté ou hépariné.
Un chiffre de plaquettes inférieur à 150 000/mm3 correspond à une thrombocytopénie. Un excès de plaquettes s'appelle une thrombocytose.
c) Dosage du facteur Willebrand
Cet examen est important. Il existe deux méthodes de dosage du facteur Willebrand :
- une méthode immunologique qui quantifie le facteur Willebrand par son antigénicité. On parle alors de mesure du vWF : Ag
- une méthode fonctionnelle qui quantifie le facteur Willebrand par son activité cofacteur de la ristocétine. La ristocétine est un antibiotique non utilisé en thérapeutique qui entraîne une agrégation des plaquettes en présence d'un facteur Willebrand. On parle de mesure du vWF : RCo.
En clinique, l'étude du "complexe Willebrand" doit comporter systématiquement un dosage de l'activité coagulante du facteur VIII (FVIII), du vWF : RCo, du vWF : Ag
d) Autres tests permettant de dépister les anomalies de l'hémostase primaire
-
Test de la consommation de la prothrombine
Ce test ancien a encore une grande utilité pratique. Il consiste à mesurer la prothrombine résiduelle dans le sérum. Il est altéré en cas de thrombopathie ou de maladie de Willebrand.
- Le temps d’occlusion plaquettaire est un test global d’hémostase primaire très sensible aux maladies de Willebrand qui peut être utilisé en dépistage.
Certains tests sont pratiquement
tombés en désuétude :
-
Signe du lacet :
Test peu reproductible et peu informatif. Une compression veineuse ou une dépression cutanée entraîne la formation de pétéchies. On parle du signe du lacet positif. Ceci peut être la traduction d'une thrombopénie, d'une thrombopathie ou d'une fragilité vasculaire.
-
Rétraction du caillot :
Lorsqu'on laisse au bain-marie un caillot sanguin dans son sérum, il se rétracte. Cette rétraction est due à la présence de plaquettes. La rétraction est insuffisante en cas de thrombopénie ou de thrombopathie.
e)
Tests spécialisés
- Etude des fonctions plaquettaires par
agrégométrie
Dans certains cas il est nécessaire pour étudier les fonctions plaquettaires d'avoir recours à des tests in-vitro qui sont du ressort du laboratoire spécialisé. Le test de référence est l'agrégométrie qui consiste à étudier les courbes d'agrégation plaquettaire en présence d'inducteurs d'agrégation : ADP, collagène, thrombine, ionophore calcique, ristocétine, acide arachidonique…
- Etude des récepteurs membranaires par
cytométrie en flux
La cytométrie en flux est une technique permettant de compter certaines cellules après les avoir marquées avec des anticorps spécifiques. Des développements modernes très intéressants sont en cours dans l'utilisation de la cytométrie pour l'étude des plaquettes.
2 - Tests explorant la coagulation (Tab. 2, Fig. 9A et 9B)
a) Temps de céphaline + activateur : TCA
Cet examen consiste à activer la voie intrinsèque de la coagulation par différentes substances : le Kaolin (TCK = Temps de Céphaline Kaolin), ou plus souvent la silice micronisée ou l'acide ellagique. Dans ce test, la céphaline est un phospholipide qui remplace les plaquettes. Le TCA n'est donc pas modifié en cas de thrombopénie ou de thrombopathie.
Chez l'adulte, la valeur normale
moyenne du TCA est de 30 à 34 sec
habituellement. On considère que le TCA est anormal lorsque
le rapport temps du malade/temps du témoin est supérieur à 1,2.
Un laboratoire doit donc toujours rendre un temps témoin pour permettre l'interprétation du test.
Chez l'enfant on admet que le Temps de Céphaline Activée est plus long : le rapport de coagulation est de 1,3.
Pour que le Temps de Céphaline Activé soit normal, il faut que les facteurs suivants soient normaux : facteur du système contact (facteur XII et XI, kininogène de haut poids moléculaire, prékallicréine), complexe anti hémophilique (facteur IX, facteur VIII), complexe de la prothrombinase (facteur X, facteur V) prothrombine (facteur II), fibrinogène (facteur I).
b)
Temps de Quick
Il consiste à mesurer le temps que met à ce former un caillot de fibrine lorsqu'on ajoute dans le plasma un excès de thromboplastine ou facteur tissulaire. Normalement le caillot se forme en 12 à 13 sec ce qui représente le Temps de Quick.
Il est habituel (et regrettable)
d'exprimer le Temps de Quick en %. Ceci s'appelle alors Taux de Prothrombine
(le terme est impropre car il ne reflète pas seulement les variations de la
prothrombine). Le Temps de Quick est
anormal si le rapport Temps de Quick du malade / Temps de Quick du témoin est
supérieur à 1,2.
Le Temps de Quick est normal lorsque les facteurs suivants sont normaux : facteur VII, facteur X, facteur V, facteur II, fibrinogène.
c) Dosage du fibrinogène
Cet examen est fait très fréquemment car les anomalies peuvent être responsables de troubles graves de la coagulation (syndrome de défibrination). Certaines anomalies sont acquises (coagulation intra-vasculaire disséminée: CIVD), d'autres sont congénitales (afibrinogénémie congénitale). Dans certains cas, on trouve aussi des fibrinogènes anormaux c'est-à-dire présents mais de faible qualité fonctionnelle: dysfibrinogénémie. Le taux normal de fibrinogène est de 2 à 4,5 g/l.
d)
Tests plus spécialisés: dosages séparés des facteurs de la coagulation
- Il est possible de doser individuellement chacun des facteurs de la coagulation:
exemple : dosage du facteur VIII, dosage du facteur IX permettant le diagnostic de l'hémophilie.
- Un examen de laboratoire fréquemment demandé est le dosage des facteurs du complexe prothrombinique. Lorsque ce dosage est demandé, le laboratoire dose les facteurs II, V , VII, X, c'est-à-dire les quatre facteurs vitamine K dépendant intervenant dans le Temps de Quick.
Cet examen est de grand intérêt dans le diagnostic des insuffisances hépato-cellulaires et des hypovitaminoses K.
e)
Méthode fonctionnelle- méthode antigénique
La quasi totalité des tests utilisés en coagulation explore les propriétés fonctionnelles des facteurs de coagulation. On mesure donc des activités. Dans certaines circonstances, notamment quand l'activité est diminuée, on est amené à doser par méthode immunologique les facteurs de coagulation. La méthode immunologique renseigne sur la présence et la quantité de facteur, mais non sur sa valeur fonctionnelle. Ceci explique que l'on définisse pour un même facteur deux valeurs différentes :
- La méthode fonctionnelle, le facteur est désigné par la lettre "c"
Exemple: facteur IX c ou facteur IX coagulant. Ce dosage peut être fait par une méthode
de coagulation: dosage chronométrique, ou à l'aide de substrat biochimique : dosage chromogénique ou colorimétrique.
- La méthode immunologique: le facteur est désigné par les lettres "Ag"
Exemple : facteur IX Ag ou facteur IX antigène.
Si un facteur est présent mais anormal (anomalie qualitative), on peut voir une discordance entre le dosage antigénique normal et un dosage fonctionnel perturbé.
f) Dosage des inhibiteurs de la coagulation
De plus en plus souvent en pathologie on est amené à doser les inhibiteurs de la coagulation pour essayer de comprendre la raison de thromboses à répétition.
Tous les inhibiteurs peuvent être dosés par méthode fonctionnelle ou antigénique : antithrombine, protéine C, protéine S, second cofacteur de l'héparine.
On peut aussi essayer de savoir si le patient réagit normalement à la protéine C activée. Ce test appelé Recherche de Résistance à la Protéine C activée s'exprime en ratio. Chez le sujet normal il est supérieur à une valeur voisine de 2,3, les normes étant en fait définies par chaque laboratoire en fonction des techniques (réactifs et automates).
g) Etude en biologie moléculaire
Le développement des techniques de biologie moléculaire a permis de mieux comprendre certains déficits de la coagulation et parfois d'en faire le diagnostic.
Les principales applications de la biologie moléculaire sont :
- La recherche de mutations responsables d'hémophilie A ou B. Ceci peut permettre le diagnostic de conductrice d'hémophilie ou un diagnostic anténatal.
- La recherche de la mutation responsable de la Résistance à la Protéine C Activé (R506Q ou facteur V Leiden).
- Le polymorphisme 20210 A sur le gène prothrombine : anomalie récemment mise en évidence considéré comme un facteur de risque de thrombose veineuse.
3 - Tests explorant la fibrinolyse
a) Temps de lyse des euglobulines (TLE ou test de Von Kaulla)
Cet examen de base permet de dépister les fibrinolyses excessives : on forme un caillot d'euglobuline. Celui-ci se lyse spontanément en deux heures. Un raccourcissement important (une demi-heure voire un quart d'heure du temps de lyse des euglobuline) témoigne d'une hyperfibrinolyse.
Il est possible aussi de dépister les hypofibrinolyses par un test dynamique appelé test de veinocclusion. Un premier prélèvement sanguin est effectué sans garrot : un deuxième prélèvement sanguin est effectué après mise en place d'un garrot laissé pendant 10 minutes. Sur chacun de ces prélèvements, avant et après veinocclusion on fait un temps de lyse des euglobulines. Normalement le garrot veineux entraîne un raccourcissement important du temps de lyse des euglobulines du fait de la libération d'activateur du plasminogène par la cellule endothéliale. Lorsqu'il y a déficit des activateurs du plasminogène, il n'y a pas de raccourcissement du temps de lyse des euglobulines.
Il est possible de doser de façon
spécifique les activateurs du plasminogène, le t-PA,
l'u-PA et les inhibiteurs (PAI-1, PAI-2).
b) Dosage du plasminogène sanguin
Ce dosage n'a pas un grand intérêt. Il semble que certains déficits en plasminogène puissent être associés à des thromboses.
c) Produit de dégradation du fibrinogène et D-Dimères
L'action de la plasmine sur la fibrine entraîne la formation de PDF (Produit de Dégradation de la Fibrine et du fibrinogène). Cet examen n'est donc pas spécifique puisqu'il ne différencie pas la dégradation du fibrinogène de celle de la fibrine. C'est la raison pour laquelle il a été remplacé par le dosage des D-Dimères : les D-Dimères sont des produits de dégradation spécifique de la fibrine. Ils sont positifs donc s'il y a activation de la coagulation et de la fibrinolyse.
Le dosage des D-Dimères est utilisé en pathologie, dans le diagnostic d'exclusion de thrombose veineuse profonde. Ils ont une très bonne valeur prédictive négative: un patient pour lequel les D-Dimères sont négatifs avec une technique sensible (ELISA) est exempte de thrombose veineuse (sensibilité >95%).
Paramètre Valeur habituelle Pathologique si
TCa 30-36 s M > 1,2 x T ( 1,3 x T chez enfant)
TQ (TP) 12-13 s (70-150%) M > 1,2 x T (1,3 x T chez enfant)
Fibrinogène 1,5-4 g/l Hors-normes
Facteurs coag 60-150% < 60% (NN: taux bas de F II, IX,X, XI et XII)
F Willebrand 60-150 % Fonction du Groupe sanguin
TS (Ivy) 4 à 8 mn > 10 mn (causes d’erreur)
TLE > 2 heures > 1h 30
ATIII, PC, PS 60-150 % Hors-normes
RPCA Ratio: 2,3 à 4 Ratio >2,3 (mutation R506Q)
D-Dimères latex Neg Positivité, exprimée en +, ++, +++
D-Dimères Elisa < 400 ng/ml Hors normes
Complexes solubles Négatifs Positivité, exprimée en +, ++, +++
Tableau 2 : Valeurs normales des tests de coagulation

Fig 9A : Schéma d’activation de la coagulation in vitro

Fig 9B : Même schéma que Fig 9A, avec les numéros de facteur
- Le temps de céphaline + activateur explore la voie intrinsèque
(partie gauche: F XII, XI, IX, VIII, X, V, II, I)
- Le temps de Quick explore la voie extrinsèque
( partie droite: F VII, X, V, II, I)
V - APPLICATIONS DES TESTS
D'HEMOSTASE
1 - Dépistage du risque hémorragique
Le dépistage du risque hémorragique ne fait pas appel aux tests d'hémostase.
L'examen le plus sensible pour dépister un risque hémorragique en particulier en pré-opératoire est l'interrogatoire qui doit être particulièrement soigneux : recherche d'antécédents hémorragiques personnels et familiaux, de signes hémorragiques même discrets : gingivorragie, ménorragie, épistaxis, ecchymose spontanée, saignement prolongé lors des piqûres ou des coupures, saignement prolongé après petit geste chirurgical ou des extractions dentaires...)
Dans certains cas l'interrogatoire n'est pas possible ou insuffisant : patient inconscient ou répondant mal aux questions, enfants...
On est alors amené à passer directement à la deuxième étape : tests biologiques pour diagnostic de syndrome hémorragique.
2 - Tests d'hémostase pour le diagnostic de syndrome hémorragique
Quatre examens de base sont nécessaires : numération plaquettaire, Temps de Quick, Temps de Céphaline + Activateur, dosage du fibrinogène.
Les principales orientations fournies par ces tests d'hémostase sont les suivantes:
- Thrombopénie
Après avoir éliminé une fausse thrombopénie, le problème est de savoir s'il s'agit d'une thrombopénie centrale (due à une anomalie de la moelle osseuse, ou périphérique: la moelle osseuse fonctionne bien mais les plaquettes sont détruites séquestrées ou consommées (cf. cours d'hématologie clinique).
- TCA normal + Temps de Quick allongé
Il s'agit habituellement d'un déficit en facteur VII qui peut être congénital ou acquis
-
TCA allongé+Temps de Quick normal
- Hémophilie A et B surtout
- Les autres anomalies responsables d'allongement du TCA et du Temps de Quick ne sont habituellement pas hémorragiques: il s'agit surtout des anomalies du système contact, et des anticoagulants circulants de type lupique (Lupus anticoagulant)

Tableau 3 : Diagnostic d’un allongement isolé du Temps de Quick (TQ)
ou du temps de céphaline +
activateur (TCA)
- TCA allongé + Temps de Quick allongé
- Anomalie du fibrinogène: un fibrinogène très bas (< à 1g/l) allonge le Temps de Quick. La baisse du fibrinogène peut être d'origine
. soit congénitale: afibrinogénémie, hypofibrinogénémie, dysfibrinogénémie
. soit acquise: CIVD, fibrinolyse aiguë
- Devant un allongement du TCA et du Temps de Quick, il faut demander le dosage des facteurs du complexe prothrombinique: ceci permet de mettre en évidence les déficits isolés en facteur II, V, VII, X, des déficits combinés présents dans les insuffisances hépato-cellulaires et les hypovitaminoses K.
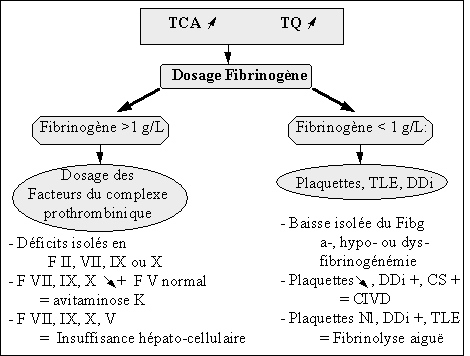
Tableau 4 : Diagnostic d’un allongement du temps de Quick
et du
temps de céphaline + activateur
- Devant un syndrome hémorragique même fruste associé à une numération plaquettaire normale, un dosage fibrinogène normal et un Temps de Céphaline Activé, un temps de Quick normaux, il faut doser le facteur Willebrand. Ce dosage doit être associé à un dosage du facteur VIII. Dans 10 à 20 % des cas de maladie de Willebrand modérée, le TCA est normal.
Si dans cette circonstance le facteur Willebrand est normal, on a recours aux examens spécialisés plaquettaires pour rechercher une anomalie fonctionnelle (test d'agrégation plaquettaire).

Tableau 5 : exploration d’un syndrome hémorragique
3 - Test d'hémostase pour bilan de thromboses veineuses récidivantes :
L’exploration doit être faite à distance du dernier épisode thrombotique et si possible en l’absence de traitement anticoagulant.
On dose alors l'antithrombine, la Protéine C, la Protéine S et on fait une recherche de résistance à la Protéine C activée et de lupus anticoagulant (éventuellement associée à la recherche d’anticorps antiphospholipides).